library(dada2, quietly = TRUE) # pipeline principal: ASVs, filtrado, errores, taxonomía
library(tidyverse, quietly = TRUE) # manipulación y visualización de datos
library(seqinr) # utilidades para secuencias FASTA
library(ShortRead) # lectura y QC de archivos FASTQ
library(fs) # operaciones con archivos y rutas
library(readr) # lectura/escritura de CSVs
set.seed(42)DADA2 — Filtrado inicial
El filtrado inicial elimina lecturas de baja calidad antes de que entren al pipeline de denoising. Es el primer paso de control de calidad específico de DADA2: define la longitud de truncamiento, elimina lecturas con demasiados errores esperados y remueve contaminantes (PhiX, bases ambiguas). Lo que entre a filterAndTrim limpio, saldrá como ASVs confiables.
Configuración del entorno
cd ~/metabarcoding-code
RDefinir rutas y parámetros
Qué hace: define el directorio de trabajo, las rutas de entrada/salida, y genera un identificador único para esta corrida basado en la fecha y hora.
setwd("~/metabarcoding-code")
fastq_location <- "for_dada2"
output_location <- "final_data"
metadata_location <- "metadata"
primer_csv <- file.path(metadata_location, "primer_data.csv")
# Crear directorios de salida
dir.create(file.path(output_location, "logs"), recursive = TRUE, showWarnings = FALSE)
dir.create(file.path(output_location, "csv_output"), recursive = TRUE, showWarnings = FALSE)
dir.create(file.path(output_location, "rdata_output"), recursive = TRUE, showWarnings = FALSE)
i <- 1 # fila de primer_data.csv a procesar
run_name <- format(Sys.time(), "%Y%m%d_%H%M")i— selecciona qué locus procesar. Si tienes varios marcadores (12S, COI, etc.), cambia este valor para cada unorun_name— prefijo único con fecha y hora (ej.20260325_1430); evita que corridas sucesivas sobreescriban los mismos archivos
Leer parámetros del locus
Qué hace: lee primer_data.csv para obtener la configuración específica del marcador: nombre del locus, longitud máxima de truncamiento, base de datos taxonómica, etc. Permite procesar diferentes marcadores con el mismo script.
primer_cols <- readr::read_csv(primer_csv, n_max = 0, show_col_types = FALSE) %>% names()
primer.data <- readr::read_csv(
primer_csv,
col_types = readr::cols(
!!primer_cols[1] := readr::col_character(),
!!primer_cols[2] := readr::col_character(),
.default = readr::col_guess()
)
)
primer.dataQué chequear: verifica que la fila i corresponde al locus que quieres analizar. El pipeline usará primer.data$locus_shorthand[i], primer.data$db_name[i], etc. en todos los pasos siguientes.
Localizar archivos FASTQ
Qué hace: busca en for_dada2/ todos los archivos FASTQ del locus seleccionado, los separa en forward (R1) y reverse (R2), y extrae el identificador de muestra del nombre del archivo.
fnFs <- grep(primer.data$locus_shorthand[i],
sort(list.files(fastq_location, pattern = "R1_001.fastq", full.names = TRUE)),
value = TRUE)
fnRs <- grep(primer.data$locus_shorthand[i],
sort(list.files(fastq_location, pattern = "R2_001.fastq", full.names = TRUE)),
value = TRUE)
sample.names <- sub(
paste0("^", primer.data$locus_shorthand[i], "-"), "",
sub("-d[0-9]+$", "",
sapply(strsplit(basename(fnFs), "_"), `[`, 1))
)
stopifnot(length(fnFs) == length(fnRs))
stopifnot(!any(duplicated(sample.names)))
length(fnFs)
head(sample.names)Qué chequear: el número total de muestras y los primeros nombres. Si el número no coincide con lo esperado, verifica que los archivos estén en for_dada2/ y que locus_shorthand en primer_data.csv sea correcto.
Preparar rutas de archivos filtrados
Qué hace: define dónde se guardarán los archivos filtrados (for_dada2/filtered/) y elimina del análisis archivos vacíos o corruptos que podrían causar errores en pasos posteriores.
filtFs <- file.path(fastq_location, "filtered", paste0(sample.names, "_F_filt.fastq"))
filtRs <- file.path(fastq_location, "filtered", paste0(sample.names, "_R_filt.fastq"))
names(filtFs) <- sample.names
names(filtRs) <- sample.names
# Eliminar archivos vacíos o corruptos
file.empty <- function(filenames) file.info(filenames)$size == 20
empty_files <- file.empty(fnFs) | file.empty(fnRs)
fnFs <- fnFs[!empty_files]
fnRs <- fnRs[!empty_files]
filtFs <- filtFs[!empty_files]
filtRs <- filtRs[!empty_files]
sample.names <- sample.names[!empty_files]Inspección de calidad
Qué hace: genera perfiles de calidad Phred por posición para las primeras muestras. Permiten decidir visualmente dónde truncar las lecturas antes de correr el filtrado.
# Gráfica 1: Forward (primeras 6 muestras)
png(filename = file.path(output_location, "logs",
paste0(run_name, "_", primer.data$locus_shorthand[i], "_quality_forward.png")),
width = 2400, height = 1600, res = 300)
plotQualityProfile(fnFs[seq_len(min(6, length(fnFs)))]) +
ggplot2::theme_minimal(base_size = 10) +
ggplot2::labs(title = "Perfiles de calidad — Forward (R1)")
dev.off()
# Gráfica 2: Reverse (primeras 6 muestras)
png(filename = file.path(output_location, "logs",
paste0(run_name, "_", primer.data$locus_shorthand[i], "_quality_reverse.png")),
width = 2400, height = 1600, res = 300)
plotQualityProfile(fnRs[seq_len(min(6, length(fnRs)))]) +
ggplot2::theme_minimal(base_size = 10) +
ggplot2::labs(title = "Perfiles de calidad — Reverse (R2)")
dev.off()
# Gráfica 3: Muestra 1 — Forward vs Reverse lado a lado
library(gridExtra)
p_f <- plotQualityProfile(fnFs[1]) +
ggplot2::theme_minimal(base_size = 10) +
ggplot2::labs(title = "Muestra 1 — Forward (R1)")
p_r <- plotQualityProfile(fnRs[1]) +
ggplot2::theme_minimal(base_size = 10) +
ggplot2::labs(title = "Muestra 1 — Reverse (R2)")
png(filename = file.path(output_location, "logs",
paste0(run_name, "_", primer.data$locus_shorthand[i], "_quality_paired.png")),
width = 2400, height = 1000, res = 300)
gridExtra::grid.arrange(p_f, p_r, ncol = 2)
dev.off()Para descargar las gráficas a tu computadora:
scp usrXX@200.23.162.240:/home/usrXX/metabarcoding-code/final_data/logs/*quality*.png ~/Downloads/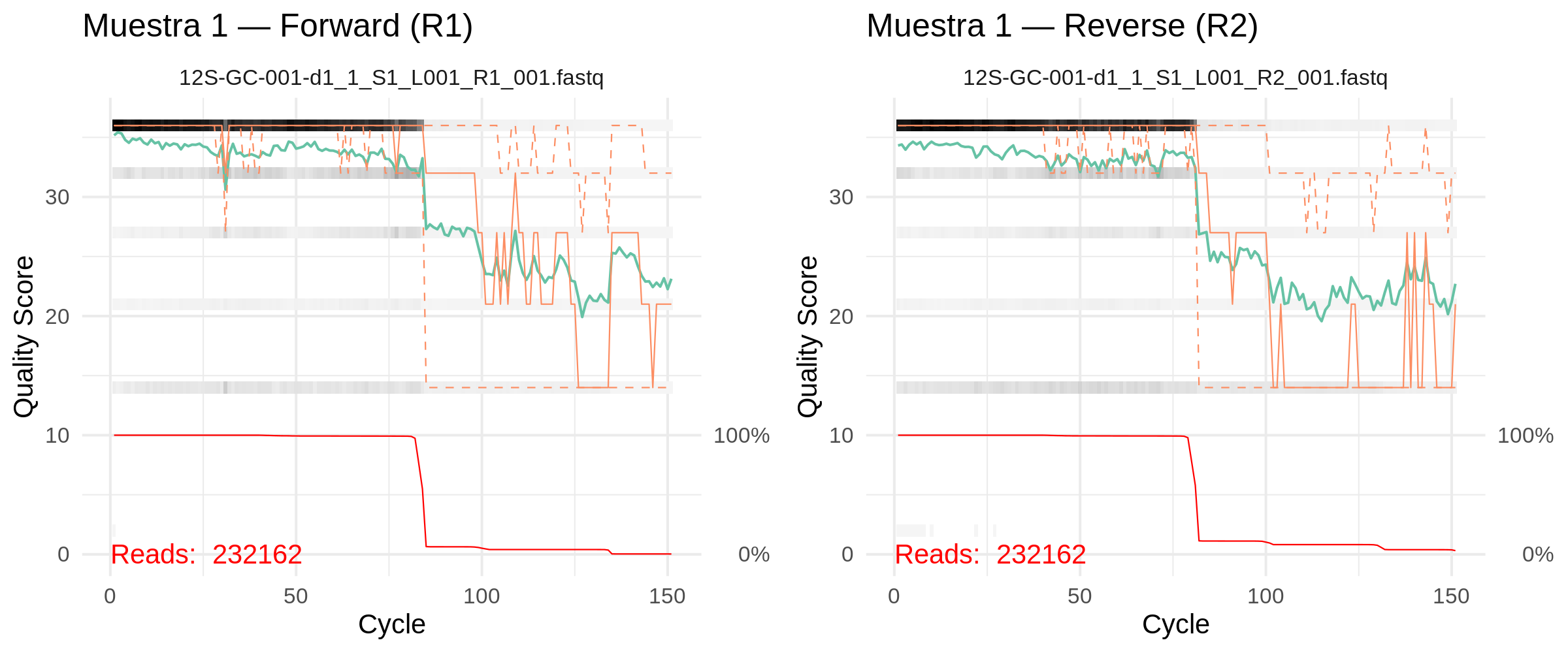
Verificación de primers
Qué hace: verifica que Cutadapt haya removido correctamente los primers. Si siguen presentes, DADA2 los interpretará como parte de la secuencia biológica y afectará el denoising.
library(Biostrings)
check_primers <- function(fastq_file, primer_seq, n = 1000) {
streamer <- ShortRead::FastqStreamer(fastq_file, n = n)
on.exit(close(streamer))
chunk <- ShortRead::yield(streamer)
seqs <- as.character(ShortRead::sread(chunk))
exact_match <- sum(grepl(paste0("^", primer_seq), seqs))
substr_seqs <- substr(seqs, 1, 50)
anywhere_match <- sum(grepl(primer_seq, substr_seqs))
list(exact = exact_match, anywhere = anywhere_match, total = length(seqs))
}
# Secuencias de primers (ajustar según tu marcador)
primer_F <- "TTAGATACCCCACTATGC" # ejemplo 12S
primer_R <- "TAGAACAGGCTCCTCTAG" # ejemplo 12S
# Verificar en R1 (forward)
check_F_in_R1 <- check_primers(fnFs[1], primer_F)
check_R_RC_in_R1 <- check_primers(fnFs[1], as.character(
Biostrings::reverseComplement(Biostrings::DNAString(primer_R))))
# Verificar en R2 (reverse)
check_R_in_R2 <- check_primers(fnRs[1], primer_R)
check_F_RC_in_R2 <- check_primers(fnRs[1], as.character(
Biostrings::reverseComplement(Biostrings::DNAString(primer_F))))
message("R1 — Primer F al inicio: ", check_F_in_R1$exact, "/", check_F_in_R1$total)
message("R1 — Primer R-RC al inicio: ", check_R_RC_in_R1$exact, "/", check_R_RC_in_R1$total)
message("R2 — Primer R al inicio: ", check_R_in_R2$exact, "/", check_R_in_R2$total)
message("R2 — Primer F-RC al inicio: ", check_F_RC_in_R2$exact, "/", check_F_RC_in_R2$total)Qué chequear: los conteos deben ser 0 o cercanos a 0. Si son altos, los primers no fueron removidos — revisa la configuración de Cutadapt o las secuencias en primer_data.csv.
Determinar el punto de truncamiento
El punto de truncamiento define hasta dónde se cortan las lecturas antes de filtrar. Depende de dos factores: la calidad Phred y la longitud real de las lecturas después de que Cutadapt removió los primers.
Paso 1 — Truncamiento por calidad (ventana deslizante)
Qué hace: recorre la calidad mediana de cada muestra con una ventana de 10 posiciones y marca dónde cae por debajo de Q30. Toma la mediana entre todas las muestras como valor global.
n <- 500000; window_size <- 10
# --- Forward ---
trimsF <- numeric(length(fnFs))
for (f in seq_along(fnFs)) {
srqa <- qa(fnFs[f], n = n)
df <- srqa[["perCycle"]]$quality
means <- rowsum(df$Score * df$Count, df$Cycle) / rowsum(df$Count, df$Cycle)
window_values <- vapply(1:(length(means) - window_size),
function(j) mean(means[j:(j + window_size)]), numeric(1))
trimsF[f] <- min(which(window_values < 30))
}
where_trim_all_Fs <- median(trimsF, na.rm = TRUE)
if (where_trim_all_Fs > primer.data$max_trim[i]) where_trim_all_Fs <- primer.data$max_trim[i]
# --- Reverse ---
trimsR <- numeric(length(fnRs))
for (r in seq_along(fnRs)) {
srqa <- qa(fnRs[r], n = n)
df <- srqa[["perCycle"]]$quality
means <- rowsum(df$Score * df$Count, df$Cycle) / rowsum(df$Count, df$Cycle)
window_values <- vapply(1:(length(means) - window_size),
function(j) mean(means[j:(j + window_size)]), numeric(1))
trimsR[r] <- min(which(window_values < 30))
}
where_trim_all_Rs <- median(trimsR, na.rm = TRUE)
if (where_trim_all_Rs > primer.data$max_trim[i]) where_trim_all_Rs <- primer.data$max_trim[i]
message("Truncamiento por calidad: F=", where_trim_all_Fs, " | R=", where_trim_all_Rs)Paso 2 — Ajuste por longitud real de lecturas
Qué hace: para amplicones cortos como 12S (~65 bp), las lecturas ya terminan antes de donde la calidad se degradaría. filterAndTrim descarta cualquier lectura más corta que truncLen, así que si truncamos a más de lo que miden las lecturas, perdemos casi todo. El pipeline calcula el percentil 5 de longitudes reales y usa el menor valor entre ese y el truncamiento por calidad.
sample_n_reads <- 100000
get_lengths <- function(fq_file, n = sample_n_reads) {
streamer <- ShortRead::FastqStreamer(fq_file, n = n)
on.exit(close(streamer))
chunk <- ShortRead::yield(streamer)
if (length(chunk) == 0) return(integer(0))
ShortRead::width(ShortRead::sread(chunk))
}
lengths_F <- purrr::map_df(fnFs, ~ {
L <- get_lengths(.x)
tibble::tibble(ReadType = "Forward", File = basename(.x), Length = L)
})
lengths_R <- purrr::map_df(fnRs, ~ {
L <- get_lengths(.x)
tibble::tibble(ReadType = "Reverse", File = basename(.x), Length = L)
})
lengths_all <- dplyr::bind_rows(lengths_F, lengths_R)p5_F <- as.integer(quantile(lengths_F$Length, 0.05, na.rm = TRUE))
p5_R <- as.integer(quantile(lengths_R$Length, 0.05, na.rm = TRUE))
where_trim_all_Fs <- min(where_trim_all_Fs, p5_F)
where_trim_all_Rs <- min(where_trim_all_Rs, p5_R)
message("Percentil 5 de longitudes: F=", p5_F, " bp | R=", p5_R, " bp")
message("Truncamiento final (calidad + distribución): F=", where_trim_all_Fs, " | R=", where_trim_all_Rs)vlines <- data.frame(
ReadType = c("Forward", "Reverse"),
xintercept = c(where_trim_all_Fs, where_trim_all_Rs)
)
ggplot(lengths_all, aes(x = Length, fill = ReadType)) +
geom_histogram(alpha = 0.6, position = "identity", binwidth = 1, color = "black") +
geom_vline(data = vlines, aes(xintercept = xintercept),
color = "red", linetype = "dashed", linewidth = 0.8) +
facet_wrap(~ ReadType, ncol = 1, scales = "free_y") +
labs(
title = "Distribución de longitudes de lecturas",
subtitle = paste0("Línea roja = truncLen (F=", where_trim_all_Fs,
" | R=", where_trim_all_Rs, ") — lecturas más cortas se descartan"),
x = "Longitud (bp)", y = "Frecuencia"
) +
theme_minimal()Qué chequear: la línea roja debe caer en la cola izquierda de la distribución, no en el centro. Si cae en el centro, estamos descartando la mayoría de las lecturas.
Filtrado y truncamiento con filterAndTrim
Qué hace: aplica simultáneamente varios filtros a cada par de lecturas y guarda solo las que pasan todos. Es el paso que más reduce el número de lecturas — pérdidas del 20–30% son normales.
out <- filterAndTrim(
fnFs, filtFs, fnRs, filtRs,
truncLen = c(where_trim_all_Fs, where_trim_all_Rs),
maxN = 0,
maxEE = c(2, 2),
truncQ = 2,
rm.phix = TRUE,
compress = TRUE,
multithread = 2,
matchIDs = TRUE
)Parámetros clave:
| Parámetro | Valor | Qué hace |
|---|---|---|
truncLen |
c(F, R) |
Trunca las lecturas a esa longitud. Lecturas más cortas se descartan |
maxN |
0 |
DADA2 requiere 0 bases ambiguas — cualquier N descarta la lectura |
maxEE |
c(2, 2) |
Máximo de errores esperados (Σ 10^(-Q/10)) por lectura. El más importante después de truncLen |
truncQ |
2 |
Trunca si hay una base con Q ≤ 2 (>60% prob. de error). Red de seguridad para bases extremas |
rm.phix |
TRUE |
Elimina lecturas del control PhiX que Illumina agrega en cada corrida |
matchIDs |
TRUE |
Si R1 pasa pero R2 no (o viceversa), descarta el par completo |
Verificar retención
pct_retained <- round(sum(out[,"reads.out"]) / sum(out[,"reads.in"]) * 100, 2)
message("Retención global: ", pct_retained, "%")
low_retention <- out[,"reads.out"] / out[,"reads.in"] < 0.3
if (any(low_retention)) warning("Muestras < 30%: ", paste(rownames(out)[low_retention], collapse=", "))
head(out)| Retención global | Interpretación |
|---|---|
| > 70% | Normal |
| 50–70% | Aceptable — revisar truncLen |
| < 50% | Bajo — aumentar maxEE a 3–5, o relajar truncLen |
| < 30% en alguna muestra | Problemática — revisar con FastQC antes de excluirla |
Guardar checkpoint
save(out, filtFs, filtRs, fnFs, fnRs, sample.names, primer.data, run_name,
fastq_location, output_location, metadata_location,
file = file.path(output_location, "rdata_output",
paste0(run_name, "_", primer.data$locus_shorthand[i],
"_checkpoint1_filterAndTrim.RData")))
message("Checkpoint 1 guardado")load(file.path(output_location, "rdata_output",
paste0(run_name, "_", primer.data$locus_shorthand[i],
"_checkpoint1_filterAndTrim.RData")))
TipPara el curso
- ¿Cuál fue el truncamiento final para Forward y Reverse? ¿Lo determinó la calidad o la distribución de longitudes?
- ¿Qué porcentaje global de lecturas pasó el filtrado? ¿Hay muestras con retención < 30%?
- En la gráfica de distribución de longitudes, ¿la línea roja cae en la cola izquierda o en el centro?
- ¿Los primers fueron removidos? ¿Los conteos de verificación son cercanos a 0?