library(dada2, quietly = TRUE)
library(tidyverse, quietly = TRUE)
library(ShortRead)
set.seed(42)Dada2 - Dereplicación e Inferencia de Errores
Este paso tiene dos partes: primero dereplicamos las lecturas (agrupamos duplicados y contamos abundancias), luego aprendemos el modelo de error específico de nuestro run para poder separar errores técnicos de variantes biológicas reales. El resultado es el conjunto de ASVs: secuencias verificadas a resolución de nucleótido único.
Configuración del entorno
Carga el checkpoint guardado al final del filtrado de calidad:
setwd("~/metabarcoding-code")
output_location <- "final_data"
metadata_location <- "metadata"
primer_csv <- file.path(metadata_location, "primer_data.csv")
primer.data <- readr::read_csv(primer_csv, show_col_types = FALSE)
i <- 1
checkpoint_derep <- list.files(file.path(output_location, "rdata_output"),
pattern = "Dereps\\.RData$", full.names = TRUE)
load(checkpoint_derep)
message("Checkpoint cargado: ", basename(checkpoint_derep))Paso 1 — Dereplicación
Qué hace: agrupa todas las lecturas con exactamente la misma secuencia en una sola entrada y registra cuántas veces apareció cada una (su abundancia). Esto reduce el volumen de datos drásticamente — en nuestros datos, de ~158,000 lecturas a ~6,000 secuencias únicas — sin perder ninguna información biológica. Los pasos siguientes trabajan con esas secuencias únicas, no con todas las lecturas repetidas.
exists <- file.exists(filtFs) & file.exists(filtRs)
filtFs <- filtFs[exists]
filtRs <- filtRs[exists]
sample.names <- sample.names[exists]
derepFs <- derepFastq(filtFs, verbose = TRUE)
derepRs <- derepFastq(filtRs, verbose = TRUE)
names(derepFs) <- sample.names
names(derepRs) <- sample.namesQué chequear: el diagnóstico de la primera muestra muestra cuántas secuencias únicas hay y cuál es el ratio de compresión:
n_unique_F <- length(derepFs[[1]]$uniques)
total_reads_F <- sum(derepFs[[1]]$uniques)
compression_ratio <- round(n_unique_F / total_reads_F * 100, 2)
message("Secuencias únicas Forward: ", n_unique_F)
message("Ratio de compresión: ", compression_ratio, "%")
message("Top 5 abundancias: ", paste(head(sort(derepFs[[1]]$uniques, decreasing = TRUE), 5), collapse = ", "))
if (compression_ratio < 1)
message("INFO: Baja diversidad (< 1% únicas). Normal en muestras con especies dominantes.")
if (compression_ratio > 50)
warning("Alta diversidad inusual (> 50% únicas). Revisa calidad del filtrado o contaminación.")| Ratio de compresión | Interpretación |
|---|---|
| < 1% | Muy pocas secuencias únicas — muestra dominada por pocas especies |
| 1–10% | Rango típico para eDNA |
| 10–30% | Alta diversidad o amplicones largos |
| > 50% | Inusual — posible contaminación o baja calidad de filtrado |
El pipeline guarda estadísticas de dereplicación para todas las muestras en final_data/logs/:
derep_stats <- data.frame(
Sample = sample.names,
Unique_seqs_F = sapply(derepFs, function(x) length(x$uniques)),
Total_reads_F = sapply(derepFs, function(x) sum(x$uniques)),
Unique_seqs_R = sapply(derepRs, function(x) length(x$uniques)),
Total_reads_R = sapply(derepRs, function(x) sum(x$uniques))
)
derep_stats$Compression_ratio_F <- with(derep_stats,
round(Unique_seqs_F / Total_reads_F * 100, 2))
derep_stats$Compression_ratio_R <- with(derep_stats,
round(Unique_seqs_R / Total_reads_R * 100, 2))
write.csv(derep_stats,
file.path(output_location, "logs",
paste0(run_name, "_", primer.data$locus_shorthand[i], "_derep_stats.csv")),
row.names = FALSE)Checkpoint — después de dereplicación
save(out, filtFs, filtRs, fnFs, fnRs, sample.names, primer.data, run_name,
fastq_location, output_location, metadata_location,
where_trim_all_Fs, where_trim_all_Rs,
derepFs, derepRs, n_unique_F, total_reads_F, compression_ratio,
file = file.path(output_location, "rdata_output",
paste0(run_name, "_", primer.data$locus_shorthand[i], "_checkpoint2_Dereps.RData")))
message("Checkpoint 2 guardado (Dereps)")Paso 2 — Aprender el modelo de error
Qué hace: estima las tasas de error específicas de tu run de secuenciación. La secuenciadora Illumina comete errores que dependen del tipo de cambio de base (A→C, A→G, etc.) y del Q-score de cada posición. learnErrors() aprende estas tasas directamente de tus datos — iterando hasta que las estimaciones estabilicen — para no usar valores genéricos que no reflejan el perfil real de tu equipo y reactivos.
¿Cómo construye la tabla de tasas?
Imagina que tienes esta lectura:
Secuencia real: A T G C A T G
Secuencia leída: A T G C C T G
↑
error — leyó C en vez de ALa secuenciadora asigna un Q-score a esa posición — digamos Q20. Eso significa 1% de probabilidad de error. DADA2 registra ese evento: “con Q20, vi un cambio A→C”.
Después de millones de bases, cuenta todos esos eventos:
A→C con Q20: ocurrió 500 veces de 50,000 posiciones = 1%
A→C con Q30: ocurrió 50 veces de 50,000 posiciones = 0.1%
A→C con Q40: ocurrió 5 veces de 50,000 posiciones = 0.01%Hace eso para los 12 cambios posibles (A→C, A→G, A→T, C→A, C→G, C→T, G→A, G→C, G→T, T→A, T→C, T→G) y para cada nivel Q0–Q40. El resultado es la tabla que ves en plotErrors — 16 paneles, uno por cada combinación incluyendo A→A, C→C, etc.
El problema del huevo y la gallina
Para contar cuántas veces ocurrió A→C necesitas saber cuál era la base real — pero no la sabes, solo tienes lo que leyó la secuenciadora. ¿El A→C que observas es un error (la base real era A) o una variante real (la base realmente es C en ese organismo)? No puedes saberlo sin el modelo de error, y no puedes tener el modelo sin saberlo. DADA2 lo resuelve iterando:
- Ronda 0: asume que TODO es error. Calcula las tasas iniciales con esa suposición
- Ronda 1: con esas tasas evalúa cada secuencia — las muy abundantes probablemente son reales, las separa. Recalcula las tasas usando solo lo que considera errores
- Rondas siguientes: repite. Cada vez la separación es más precisa y las tasas más confiables
- Convergencia: cuando las tasas de la ronda N y N+1 son prácticamente idénticas, el modelo dejó de cambiar
En nuestros datos Forward convergió en 9 rondas, Reverse en 14 — Reverse tarda más porque sus lecturas tienen mayor tasa de error y el modelo necesita más iteraciones para estabilizarse.
Antes de lanzarlo, el pipeline valida que haya suficientes datos:
nbases_target <- 1e8 # 100 millones de bases (aumentar si tienes > 50 muestras)
max_consist <- 25 # más iteraciones que el default (10) para forzar convergencia
# Validar datos disponibles
total_bases_F <- sum(sapply(filtFs, function(f) if (file.exists(f)) file.info(f)$size else 0))
total_bases_R <- sum(sapply(filtRs, function(r) if (file.exists(r)) file.info(r)$size else 0))
message("Forward disponible: ~", round(total_bases_F / 1e6, 1), " MB")
message("Reverse disponible: ~", round(total_bases_R / 1e6, 1), " MB")
if (total_bases_F < 1e6 || total_bases_R < 1e6)
warning("Datos muy limitados (< 1 MB). El modelo puede ser inestable.")tryCatch() es un envoltorio de seguridad: ejecuta el código dentro y, si ocurre un error, lo captura y muestra un mensaje claro en lugar de que R falle silenciosamente. Aquí lo usamos porque learnErrors() puede tardar varios minutos — si falla a mitad, queremos saber exactamente por qué.
errF <- tryCatch({
learnErrors(filtFs, nbases = nbases_target, randomize = TRUE,
multithread = 2, MAX_CONSIST = max_consist, verbose = TRUE)
}, error = function(e) {
stop("Error al aprender modelo Forward: ", conditionMessage(e))
})
errR <- tryCatch({
learnErrors(filtRs, nbases = nbases_target, randomize = TRUE,
multithread = 2, MAX_CONSIST = max_consist, verbose = TRUE)
}, error = function(e) {
stop("Error al aprender modelo Reverse: ", conditionMessage(e))
})Parámetros clave:
| Parámetro | Valor | Qué hace |
|---|---|---|
nbases |
1e8 |
Bases a usar para aprender el modelo. DADA2 toma muestras hasta alcanzar ese total |
randomize |
TRUE |
Selecciona lecturas al azar — produce un modelo más representativo |
multithread |
2 |
Núcleos a usar (limitado a 2 por el servidor compartido) |
MAX_CONSIST |
25 |
Iteraciones máximas. El default es 10; 25 garantiza convergencia en datos complejos |
Qué chequear en la salida:
103158594 total bases in 1637438 reads from 20 samples will be used for learning the error rates.
selfConsist step 1 ..........
selfConsist step 2
...
Convergence after 12 rounds.- Número de bases y muestras usadas — confirma que DADA2 alcanzó
nbases_target - Convergencia — cuántas rondas tardó en estabilizar las estimaciones
| Rondas hasta convergencia | Interpretación |
|---|---|
| 3–5 | Convergencia rápida — datos de buena calidad |
| 5–15 | Rango típico |
| 15–25 | Datos complejos o ruidosos — el modelo es válido |
| No converge (llega a MAX_CONSIST) | Aumenta MAX_CONSIST a 50 o revisa la calidad del filtrado |
Después del aprendizaje, el pipeline valida que las tasas estimadas sean razonables:
# Validar modelo Forward
if (!is.null(errF$err_out)) {
rn <- rownames(errF$err_out)
error_rows <- rn[substr(rn, 1, 1) != substr(rn, 3, 3)] # A2C, A2G, etc.
q_cols <- as.integer(colnames(errF$err_out))
hq_cols <- which(q_cols >= 20) # columnas Q≥20
err_hq <- errF$err_out[error_rows, hq_cols, drop = FALSE]
max_err <- max(err_hq, na.rm = TRUE)
message("Tipos de transición de error: ", length(error_rows), " (de 16 posibles)")
message("Tasa de error máxima en Q≥20: ", round(max_err, 5),
" (típico < 0.05; verifica con plotErrors() si > 0.10)")
if (max_err > 0.1)
message("Tasa de error > 10% en Q≥20. Verifica con plotErrors() que el modelo ajuste bien los datos.")
else
message("Modelo de error OK.")
}| Tasa máxima en Q≥20 | Interpretación |
|---|---|
| < 1% | Excelente |
| 1–5% | Normal para Illumina |
| 5–10% | Marginal — revisa los gráficos |
| > 10% | Problemático — revisa calidad del run o el filtrado |
Paso 3 — Visualizar el modelo de error
Qué hace: genera gráficas con las 16 posibles transiciones entre bases (A→A, A→C, A→G, …, T→T). Permiten confirmar visualmente que el modelo aprendido es coherente con los datos.
png(file.path(output_location, "logs",
paste0(run_name, "_", primer.data$locus_shorthand[i], "_errF.png")),
width = 1200, height = 1200, res = 150)
print(plotErrors(errF, nominalQ = TRUE))
dev.off()
png(file.path(output_location, "logs",
paste0(run_name, "_", primer.data$locus_shorthand[i], "_errR.png")),
width = 1200, height = 1200, res = 150)
print(plotErrors(errR, nominalQ = TRUE))
dev.off()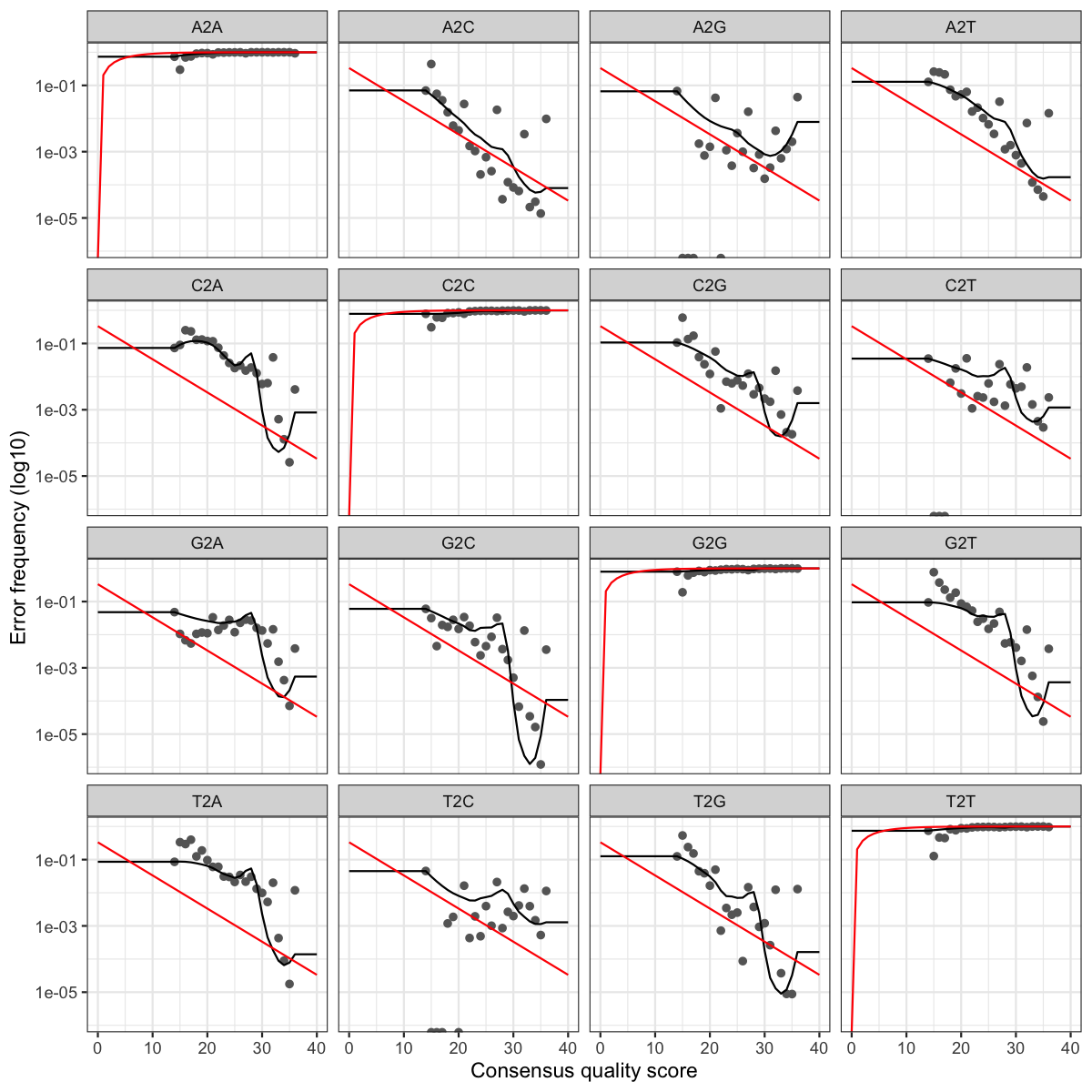
Cómo leer la gráfica:
16 paneles organizados en cuadrícula 4×4 — la fila es la base real (A, C, G, T) y la columna es la base leída. Tres elementos en cada panel:
- Puntos grises — tasas de error observadas en tus datos, agrupadas por Q-score
- Línea negra — el modelo aprendido por DADA2 (lo que usará en el denoising)
- Línea roja — tasa teórica nominal del Q-score (lo que Illumina promete)
Paneles diagonales (A2A, C2C, G2G, T2T): la base se leyó correctamente — frecuencia de error ~0.99, la línea roja sube bruscamente a 1.0. No tienen información útil sobre errores.
Paneles fuera de la diagonal — aquí está la información. Un modelo bien ajustado muestra tres cosas:
- La línea negra baja de izquierda a derecha — a mayor Q-score, menos error
- La línea negra sigue los puntos grises — el modelo se ajustó bien a los datos reales
- La línea negra está por encima de la línea roja — las tasas reales son mayores que las teóricas, especialmente en Q altos. Esto es normal — la definición nominal del Q-score es optimista, y por eso DADA2 aprende su propio modelo en lugar de usar las tasas teóricas
Oscilaciones en la línea negra (como en G2C alrededor de Q30): indican pocos datos en esos bins de calidad. No es un problema si el patrón general sigue siendo decreciente.
Checkpoint — después de learnErrors
save(errF, errR, derepFs, derepRs, filtFs, filtRs, sample.names, primer.data, run_name, out,
file = file.path(output_location, "rdata_output",
paste0(run_name, "_", primer.data$locus_shorthand[i],
"_checkpoint2_learnErrors.RData")))
message("Checkpoint 2 guardado (learnErrors)")load(file.path(output_location, "rdata_output",
paste0(run_name, "_", primer.data$locus_shorthand[i], "_checkpoint2_learnErrors.RData")))Paso 4 — Inferencia de ASVs con dada()
Qué hace: este es el paso central de DADA2. Toma las secuencias dereplicadas y el modelo de error aprendido, y determina cuáles secuencias únicas son variantes biológicas reales (ASVs) y cuáles son errores de secuenciación. Si la diferencia entre dos secuencias es consistente con un error según el modelo, las agrupa; si no puede explicarse como error, las mantiene separadas como ASVs distintas.
dadaFs <- dada(derepFs, err = errF, multithread = 2)
dadaRs <- dada(derepRs, err = errR, multithread = 2)# Resumen de la primera muestra
dadaFs[[1]]Qué chequear:
dada-class: object describing DADA2 denoising results
140 sequence variants were inferred from 6374 input unique sequences.De 6,374 secuencias únicas (dereplicación), DADA2 determinó que solo 140 son variantes biológicas reales. Las otras ~6,234 son errores de secuenciación colapsados de vuelta a su ASV de origen.
¿Cómo decide cuál es real y cuál es error? Para cada secuencia poco abundante calcula: ¿cuál es la probabilidad de ver esta secuencia por azar, solo por errores de secuenciación? Si esa probabilidad es menor que OMEGA_A = 1e-40 — es decir, es tan improbable que sea un error — la declara variante real aunque aparezca pocas veces. 1e-40 es un umbral extremadamente estricto (1 en 10⁴⁰), lo que hace que DADA2 sea muy conservador: prefiere perder una variante rara real antes que incluir un error.
| ASVs / secuencias únicas | Interpretación |
|---|---|
| < 5% | Normal para eDNA de peces con 12S — amplicón corto, pocas especies |
| 5–15% | Alta diversidad o amplicón más largo |
| > 50% | Inusual — revisa el modelo de error o la calidad del filtrado |
Checkpoint — después de dada()
save(dadaFs, dadaRs, derepFs, derepRs, errF, errR, sample.names, primer.data, run_name, out,
file = file.path(output_location, "rdata_output",
paste0(run_name, "_", primer.data$locus_shorthand[i],
"_checkpoint3_dada.RData")))
message("Checkpoint 3 guardado (dada)")load(file.path(output_location, "rdata_output",
paste0(run_name, "_", primer.data$locus_shorthand[i], "_checkpoint3_dada.RData")))
TipPara el curso
- ¿Cuál es el ratio de compresión de la dereplicación en tu muestra? ¿Coincide con el rango esperado?
- ¿En cuántas rondas convergió el modelo Forward? ¿Y Reverse?
- Revisa
plotErrors()— ¿la línea negra sigue los puntos grises en todos los paneles? - ¿Cuántas ASVs se infirieron en la primera muestra? ¿Qué porcentaje de las secuencias únicas representan?
→ Siguiente: DADA2 — Remoción de quimeras y seguimiento de lecturas